
Abstract
Biopsy information and protein Prostate-Specific Antigen (PSA) levels are the most robust information available to oncologists worldwide to diagnose and decide therapies for prostate cancer patients. However, prostate cancer presents a high risk of recurrence, and the technologies used to evaluate it demand more complex resources. This paper aims to predict Biochemical Recurrence (BCR) based on Whole Slide Images (WSI) of biopsies, Gleason scores, and PSA levels. A U-net model was used to segment phenotypic features and trained on images from the Prostate Cancer Grade Assessment (PANDA) database to segment tumorous regions from pre-processed and scored WSI of biopsies. Then, the model was tested on data from publicly available repositories achieving an Intersection over Union of 87%. Tissue features, Gleason scores, and PSA levels provided high accuracy and precision in classifying patients according to their risk of presenting recurrence, for any Gleason score sampled. The trained classifier model demonstrated a 79.2% relative accuracy, and a precision of 69.7% for patients experiencing recurrences before 24 months. Our results provide a robust, cost-efficient approach using already available information to predict the risk of BCR.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Prostate cancer diagnosis and treatment strategies are based on the architecture of tissue structures such as the Gleason score, and the blood levels of the protein Prostate-Specific Antigen (PSA). Biopsies and PSA levels are reliable technologies for oncologists due to their clinical utility [1]. Alternatives with higher specificity based on RNA biomarkers have emerged [2] but are expensive and require a temperature-controlled supply chain of reagents, specialized equipment, and skilled personnel.
Monitoring after therapy and during remission reveals that about 30% of men will experience biochemical recurrence (BCR) with diverse response approaches available [3]. However, in Latin America (LA), access to pathologists remains challenging [4], which hinders the early detection of recurrence increases the overall mortality rate, and broadens the unequal access to cancer screening and follow-up [5]. Proposed molecular strategies [6] and investigations related to MRI images and Quantitative Ultrasound to predict clinical outcomes in prostate cancer patients [7, 8] offer valuable insights, yet their clinical implementation remains limited. The use of Whole Slide Imaging (WSI) to predict breast cancer recurrence has shown promising results [9], with an area under the receiver operating characteristic curve of 0.776, emphasizing the importance of using histological images for recurrence prediction in other cancers were glandular tissue structures are critical.
Therefore, we evaluated using Hematoxylin and Eosin (H&E) stained WSI from biopsies—the time-tested and cost-effective technology for characterizing solid tumors—to predict BCR in prostate cancer. The emergence of digital pathology has harnessed advanced artificial intelligence (AI) methods to analyze histological images, offering opportunities for enhanced diagnostic capabilities [10]. Given the shortage of experienced healthcare providers, cancer control in limited resource areas is overall inadequate and deficient [11]. Integrating AI methods into medical practices can improve personalized medicine by boosting clinical evidence and patient-clinician interaction in regions with limited resources [12].
Artificial Neural Networks (ANN) are commonly used for medical imaging segmentation research. For instance, Noorbakhsh et al. [13] trained a Convolutional Neural Network (CNN) model on 27,815 histological slides from 23 cancer types to highlight shared and distinct spatial behaviors within tumors across different cancers. Xu et al. [14] proposed an algorithm that segments glands in histological images using three CNN-based channels: Foreground–background segmentation, gland boundary detection, and gland localization. However, segmenting prostate glands has always been challenging due to their inherent heterogeneity and notable variation in appearance. Ren et al. [15] employed a CNN model with a superpixel segmentation method and obtained a glandular segmentation accuracy of 0.83. Nonetheless, these approaches only work well on clearly defined gland contours and single lumina. To address these issues, Salvi et al. [16] combined active contour and CNN to segment prostate glands in histological images, achieving a precision of 0.91, and detecting prostate glands with discontinuous and absent lumina. In addition, Qiu et al. [17] developed a pyramid semantic parsing network. Still, it was focused predominantly on a binary classification between non-tumorous and tumorous regions and performed poorly on contiguous glands or clusters of nuclei. Therefore, a model capable of validation using diverse databases, with a large dataset for robust training and testing, is needed to avoid overfitting and achieve an efficiently trained model.
Recently, deep learning models have been used to predict the presence of biomarkers, such as gene expression status, within segmented tissues of interest in histological images. These models also predict patient responses to treatments [18] and even their survival probability after surgery [19]. Nonetheless, most prognostic and diagnostic approaches only focus on final accuracy scores, neglecting the morphological features responsible for clinical manifestations. Foroughi pour et al. showed the relevance of correlating H&E-derived CNN features to biological phenotypes, whether these features are specific to individual cancer types, or multiple cancers, especially in related tissue types [20].
Previous attempts to use WSI to predict the biochemical recurrence in prostate cancer focused on patients with high Gleason scores [21,22,23] but did not address the need to include and understand the role of morphological features in the recurrence. Certain methods [24] emphasize the anatomical features responsible for the recurrence through a CNN; however, the complex structure makes it unsuitable for clinical use in institutions with limited computational resources.
In this study, phenotype characteristics including morphology and spatial arrangement of prostate glands, are collected from histological images, through an accessible and easy-to-operate U-net model. Then, these characteristics are processed to create a classifier for patient’s stratification based on their risk of recurrence by investigating informative regions within class labels. We propose using WSI regions of interest and a cost-effective deep learning model to segment lumen, glands, and nuclei, as well as their respective features, from low Gleason to more disorganized phenotypes, reflecting that higher Gleason scores correspond to less organized glandular structures.
2 Materials and methods
2.1 Dataset
The deep learning model U-net was trained and tested with whole slide H&E-stained prostate pathology images from two distinct sources. The training set was constructed from prostate biopsies obtained from the Prostate Cancer Grade Assessment (PANDA) Challenge (https://www.kaggle.com/competitions/prostate-cancer-grade-assessment), hosted by Kaggle Inc., and accepted for the MICCAI 2020 conference. Diagnostic slides for the testing set were sourced from The Cancer Genome Atlas (TCGA) web portal (https://www.cancer.gov/tcga). In this study, 800 WSI were employed for the training process, while 500 WSI for testing. Within the TCGA dataset, each WSI was accompanied by Gleason pattern masks and the ground truths Gleason scores were also provided by TCGA based on patient reports. WSI images were equipped with corresponding masks graded by multiple pathologists and associated Gleason scores from the PANDA dataset. The training dataset included 200 patients graded with a Gleason score of 6, 200 with a Gleason score of 7, 150 with a Gleason score of 8, 150 with a Gleason score of 9, and 100 with a Gleason score of 10. Most of the WSI in the PANDA dataset correspond to patients with Gleason 6 and 7. However, when building the model, it was necessary to have a balanced dataset to warrant the reproducibility of the model. Therefore, we picked WSI including 200 patients graded with a Gleason score of 6, 200 with a Gleason score of 7, 150 with a Gleason score of 8, 150 with a Gleason score of 9, and 100 with a Gleason score of 10. A complete list of the data files used from the PANDA dataset is detailed in Supplementary Data. The patients are selected randomly according to their Gleason score, where one patient coincides with one WSI. The testing dataset consisted of the 500 WSI from the TCGA web portal (Table 1). In addition to these datasets, another dataset comprising 110 H&E-stained images consisting solely of nuclei, from the 2018 Data Science Bowl Competition (https://www.kaggle.com/c/data-science-bowl-2018), was used as an isolated experiment in the training process to verify epithelial cell segmentation. Furthermore, as a reference standard, ground truths were obtained from an experienced oncologist, outlining glands, lumen, and nuclei for 230 tiles varying from Gleason 6 to 10. The data and images downloaded from public databases are available in Supplementary Materials.
The subsequent BCR classification tasks were trained and tested on 150 patients only from the TCGA dataset. Amongst the 500 TCGA patients, 99 patients present BCR, from whom 71 with the selected genes. To balance the set, 79 patients without BCR were randomly chosen with similar disease-free time. The ensuing set is divided into 10 k-fold cross-validations.
2.2 Whole slide image preprocessing
The average file size of a WSI exceeds 1 GB, primarily due to its large image size. To address this, we utilize the process from Deron Eriksson’s Github repository “python-wsi-preprocessing” (https://github.com/deroneriksson/python-wsi-preprocessing), initially developed for the TUPAC16 challenge [25]. First, the image size is reduced by dividing the height and width by 32. Starting with an initial image size, for example, 85656x71305 pixels, we obtain a scaled-down image of 2676x2228 pixels. Additionally, to not hinder the detection of phenotypic features, disturbances like pen-marked annotations are removed. The data undergoes a combination of filters to isolate the tissue and discard the background, like tissue segmentation filtering.
To deal with the domain shift between different datasets, a histogram equalization algorithm is applied to adjust the contrast between the distinct sets and staining variability. Then, the filtered slides are split into tiles, and the same tiles are resized. Once sliced, a score is assigned to each tile according to color characteristics and tissue percentage to select the tiles with the highest score for analysis. The scoring formula considered the tissue percentage, the saturation value, the ratio between hematoxylin and eosin staining, and the quantity of tissue (to give more weight to tiles with more tissue). This score is then compared by superposing the obtained scoring heatmap with the PANDA masks (Figure 1). To keep only the tiles with tumorous tissue, five tiles were retrieved by WSI/patient with a size of 2048x2048 pixels, to enclose most segmented carcinoma while discarding background and stroma, which could otherwise add noise to the diagnosis process.

Tiles retrieved from the same image. a Whole Slide Image from PANDA set. b Corresponding mask from the PANDA set highlighting the tumorous region. c Tiles scoring heatmap from scoring formula with the overlaid mask. High tissue percentages (≥ 80%) are shown with green tiles, medium tissue percentages (≥ 10% and < 80%) with yellow tiles, low tissue percentages (> 0% and < 10%) with orange tiles, and no tissue (0%) is represented with red tiles
Variations in the staining process or digital scanning can affect the color intensities of the images and subsequently cause stain disparity between the tiles, potentially affecting future segmentation results. The collected tiles were color-normalized using the Reinhard method from the HistomicsTK library to prevent artifacts. This method transforms the color characteristics of each tile into a specific grade or standard defined by the mean and standard deviation of the target image. By applying the color conditions from the target image (displayed in Figure 2) to a source image (tile), a normalized image is produced, as depicted in Figure 2.

Color staining variance and normalization architecture
2.3 Application of the U-net model to segment phenotypic components from H&E images
We adopted the U-net model proposed by Ronneberger et al. [26] (Figure 3). This model provides a high-speed and high-accuracy method suitable for detecting and enhancing organized and disorganized tissue structure. These sets (after the preprocessing step) are transferred into Hierarchical Data Format files (HDF5) since they can efficiently support complex and heterogeneous data.

U-net architecture to segment phenotypic components from WSI
The U-Net model architecture, as depicted in Figure 1, comprises encoder and decoder paths. It initiates with a channel depth of 32 at the first level and progressively doubles this depth in five subsequent stages, ultimately reaching 1024 at the bottom level. This channel depth is then reduced back to 32 in the decoder part.
The model consists of six consecutive levels in each path, featuring a sequence of operations: 3 × 3 convolutions (represented by green arrows) followed by rectified linear units (ReLU), 2 × 2 max pooling (red arrows), up-convolution (light blue arrows), and feature concatenation (light yellow rectangles). The last layer employs a 1 × 1 convolution (purple arrow) to generate a single feature map representing the prediction results. Because of the large image sizes of 2048 × 2048 pixels, six levels were chosen in the U-Net model to avoid overlooking small phenotype elements by the architecture.
The model was implemented with an SGD optimizer, a learning rate of 0.1, a weight decay of 0.0005, and a momentum of 0.9. For each model, datasets were shuffled and divided into 90% training and 10% testing. During the training and testing phases, data augmentation was performed by horizontal and vertical flips, random cropping, and rotation. The model was evaluated during the training and testing phase with Binary Cross Entropy, tracing incorrect labeling of the data label, accuracy, and Binary Intersection over Union (IoU) metrics. The IoU was calculated using the formula: IoU = true − positives/(true − positives + false − positives + false − negatives).
Also, the segmentation results were compared to contours defined by an experienced oncologist. This involved quantifying false positive (FP), true positive (TP), false negative (FN), and true negative (TN) outcomes concerning the ground truth contour. True positives (TP) corresponded to areas segmented as glandular by our approach, which were also considered glandular in the ground truth. True negatives (TN) were regions labeled as background by our method and the ground truth. Conversely, false negatives (FN) covered areas not segmented by our approach but designated as part of the gland by the ground truth. False positives (FP) indicated regions identified as glands solely by our approach. The segmentation accuracy was quantified using the IoU, with a value of 1 representing a perfect match between the ground truth and our model’s predictions. The model was trained for two days on a laptop with an Intel Core i5 processor.
2.3.1 Segmentation of the glands
The U-net model underwent 100 training epochs, using the 4000 tiles from the 800 WSI and their matching masks, stored as HDF5 files. Individual tiles are associated with their corresponding BCR label and Gleason score. Consequently, this model can identify glands across different cancer stages, ranging from Gleason score 6 to Gleason score 10, with an accuracy of 85% when evaluated on 2500 tiles from the TCGA testing set. Figure 4 illustrates an example of the segmentation results. Ground truth masks were compared with predicted masks using a confusion matrix. From their dispositions, prostate glands can be displayed in adjacency, and won’t be distinguished independently by the model. Glands will then be reconstructed from the suspected lumen and by color intensity.

Results from U-net training and testing. a Segmentation output. b Associated predicted mask
2.3.2 Segmentation of the nuclei
The same U-net architecture was used to segment nuclei in the small dataset of 110 images (Table 1). The model was trained on 60 H&E-stained images and validated with 50 histological samples. Data augmentation was applied to a subset of the training samples, including rotation, shifting, and deformation using a displacement vector [26]. The displacement result was then sampled by Gaussian distribution and computed using bicubic interpolation. The U-net model, trained to identify and segment the nuclei, was tested on H&E-stained and solely Hematoxylin-stained images (Figure 5). The Hematoxylin-stained images were generated through a color deconvolution process applied to each of the 110 H&E-stained images. Although the Hematoxylin model exhibits a slightly lower accuracy of 76% compared to the 83% accuracy of the H&E model, it emphasizes the segmentation of darker stained nuclei.

Nuclei segmentation outputs were obtained using the trained U-Net model. a H&E-stained tiles. b U-net segmentation output for H&E-stained tiles. c Hematoxylin-stained tiles. d U-net segmentation output for Hematoxylin-stained tiles. e Adjacent nuclei are segmented as a single object. f Masks of the adjacent nuclei are segmented as a single object. g Separated nuclei resulting from the watershed algorithm
While these results are moderately accurate, subsequent processes like gland separation and reconstruction depend on precise nuclei segmentation. Recognizing this, the focus turns to resolving merged nuclei, involving the separation of nuclei recognized as one (Figure 5). A Distance Transform Watershed approach [27] was used to segment round objects like nuclei. The outcomes of this process, illustrating the successful separation of previously merged nuclei, and indicating a reduction in nuclei area, are shown in Figure 5. Nonetheless, due to the relatively stable nuclei size, nuclei density can be calculated based on the number of instances within the slide.
2.3.2.1 Detection of the lumina
Lumina differs from the rest of the carcinoma based on color characteristics. Nonetheless, the previously mentioned datasets do not contain slides with detailed lumen contours as seen in Figure 6. Conditions are applied to the post-model segmented glands to highlight the lumina while discarding artifacts with similar color characteristics.

Detection of edge lumen. a Gland segmented from the U-net model without lumen. b Convex hull subtraction consisting of stroma and lumen. c Final gland segmentation with lumen
The U-net model was trained to detect the lumen inside the glands, by considering them part of the background and creating a new contour inside the glands. The location of the lumen is crucial when distinguishing the lumen from the background. By using the hierarchy and contour tools from the Open Computer Vision library, it was possible to extract the lumina. External and internal contours were extracted from the U-net output. Then, when talking about a gland with a lumen, the presence of internal contours also called child contour is required, while the outer contour is known as the parent contour. The hierarchy of the glands is expressed according to forms like if the contour has or has not internal/child contour, or if the contour has an external/parent contour.
While looping through the contours, only those with a parent contour are considered as lumen, provided that the region size is greater than 100 pixels and, the ratio of the number of pixels with intensity over 180 (from an intensity range of 0 to 255) to the total number of pixels exceeds 0.65. For lumina at the edges, their contours were not considered child contours (Figure 6). Instead, an external envelope known as Convex Hull, was implemented to simultaneously cover the gland and prospective lumen. While creating the Convex Hull, some stroma was incorporated into the envelope. To retain only the lumen and discard stroma, the convex hull and the actual glands were subtracted (Figure 6). Then, the same aforementioned size and color conditions were applied to retain the lumen, resulting in a final gland segmentation with lumen (Figure 6).
In tiles with a high Gleason score, distinguishing glands becomes challenging. The primary indicator of a gland is the presence of cytoplasm between the lumen and the layer of nuclei, so the contour should be detected, otherwise the lumen should be discarded. To detect the homogeneity and dissimilarity of the tissue around the lumen, it is important to consider cases where the cytoplasm layer is too thin to extract this information. To address this, the segmented nuclei result from the U-net model was subtracted from the gland segmented result (Figure 7). The Euclidean distance transform was then calculated using the results of the subtraction. Bright pixels in the distance transform corresponded to the central regions of the glands when nuclei were organized as an external layer, while darker pixels indicated a nuclear region. Pixels with an intensity above 0.8 in the normalized distance map were kept and compared to the detected lumen. If the regions labeled as lumen by the distance transform fitted entirely inside the lumen identified previously, the lumen was retained; otherwise, it was discarded.

Final lumen classification from distance transformation. a The region of interest is classified as lumen from size and color. b Gland segmentation results with subtracted nuclei segmentation results. c Distance transformation. d The actual area labeled as lumen from distance transform
In Figure 7, only one region met the distance transformation requirement but did not match any lumen detected via size and shape conditions, leading to its disposal. No lumen was identified in this area, so the presence of a cluster of nuclei was assumed.
2.4 Reconstruction of the glands from the lumina
Following the steps undertaken, nuclei, glands, and lumen have been properly detected and segmented. However, in some cases, closely situated irregular glands are erroneously considered a single unit due to their proximity. Therefore, a reconstruction process was initiated to separate these merged glands. Before reconstruction, the stroma within the gland’s region was removed from the RGB images (tiles). This was accomplished by applying a precise color constraint within the HSV color space, focusing solely on color intensity while disregarding color components. The image was then converted to the HSV color space. Next, the nuclei and lumina were subtracted, and pixels in the second channel of the image with color intensity above 0.35, indicating stromal presence, were deleted. A mask with filtered stroma was generated and applied to the RGB color space images (Figure 8).

Reconstruction of the glands from detected lumen via Geodesic Active contour. a Filtered Image in RGB color space. b Dilation and erosion of RGB image with filtered stroma. c Active contour with 0.25 threshold
Assuming that glands are organized around a lumen followed by a layer of nuclei, the Delaunay triangulation [28] method was employed to link the center of the perceived lumen to the center of the nuclei detected on the image. Only the nuclei directly connected to the center of the lumen were retained and considered as the external layer of the gland. Importantly, the Delaunay triangulation required a starting point distinct from the lumen’s center. In this context, multiple points, the corners of the lumen, served as points of departure of the Delaunay. These lumen corners were detected using the Harris Corner Detection algorithm [29].
To enhance the accuracy of gland reconstruction, the slides were segmented based on their pixel values via Simple Linear Iterative Clustering (SLIC) [30]. SLIC clusters pixels with similar intensity and proximity in the image, so SLIC segmentation was performed with 1400 segments. Glands were reconstructed via Geodesic Active Contour, involving an evaluation of pixel intensity surrounding the lumen. This method requires an initial reference point to construct a closed contour for the region of interest. Commencing from the lumen, the contour extended until the pixel color intensity matched the nuclei's, thereby establishing the gland boundaries based on their nuclei layer, which served as the stopping threshold for the active contour.
Given significant intensity variations in biopsy images, potentially affecting the final detected contour by the geodesic model, the filtered RGB tiles were dilated and eroded, to enhance edge contours while smoothing internal pixels (Figure 8). Subsequently, these treated tiles were processed with a Geodesic Active Contour of 0.25 threshold. Nonetheless, as these contours were computed based on the dilated and eroded representations of the image, the final contour on the unaltered RGB slide might not fully encompass the nuclei layer (Figure 8). The active contour edge was superposed onto the pre-processed SLIC segmentation to address this. The average color intensity of the SLIC region was retrieved, and segments with an average pixel intensity below 0.20, corresponding to the color of stained nuclei, and crossed through by the active contour, were retained and integrated into the final area of interest.
2.5 Feature extraction and classification
After gland reconstruction, 37 features were extracted, as presented in Table 2. These features encompass various aspects of gland architecture conventionally associated with prostate cancer. Additionally, supplementary phenotype components were collected, including features related to lumen quality, structural attributes, tumor volume, nuclei propagation, and stroma quantification. Each feature was discretized into five intervals to make these features compatible with our classification models. We also included the Gleason score in our feature set.
These features were fed into three classifiers -Support Vector Machine, logistic regression, and decision tree- to predict BCR and relapse time. The classifiers were selected over more recent deep-learning models for their high interpretability and transparency in classification tasks. Additionally, their ability to be integrated in clinical settings, while operating on standard hardware, and maintaining high performance, made them the preferred choice. The models were evaluated using accuracy and precision. Accuracy (TruePositives+TrueNegatives)/(TruePositives+TrueNegatives+FalsePositives+FalseNegatives) measures the overall model performance, while precision (TruePositives/(TruePositives + FalsePositives) assesses the model’s ability to make precise positive predictions.
3 Results
The juxtaposition of the ground truth from a medical expert and the alleged segmentation results, visualized in Fig. 9, yielded a precision rate of 81% across 46 patients (230 tiles) and an Intersection over Union (IoU) of 0.87. This confusion matrix visualization is derived from the Morphological Geodesic Active Contours from the corner of the lumen superposed with SLIC 1400 segments.

Confusion matrix visualization with Morphological Geodesic Active Contours from lumen. Color coding employed: magenta for False Positives (FP), yellow for False Negatives (FN), cyan for True Positives (TP), and black for True Negatives (TN)
Now, shifting our focus to classifier results, the precision of the 3 selected classifiers was compared utilizing extracted features alongside the Gleason score (Table 3). Regarding BCR prediction, the logistic regression model excels with a classification average precision of 75%, to predict whereas the patients according to their histological attributes will present a risk of BCR. The SVM and decision tree models achieve 68% and 67.5% average precision, respectively. Consequently, logistic regression stands out as the most effective classifier for BCR prediction, correctly classifying 169 out of 225 tiles with 75.2% accuracy.
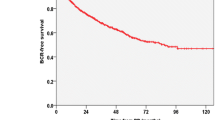
To improve overall survival, our analysis delves deeper, stratifying patients with BCR into two equal groups: those experiencing BCR within 24 months and after 24 months. The 24-month cut-off is a statistical necessity driven by the dataset’s constraints. Balancing the dataset ensured that the model could train effectively, given the limited recurrence cases while reducing class imbalance bias and optimizing the model's ability to generalize predictions across the limited dataset.
In these subgroups, precision results for each classifier are shown in Table 3. As before, the logistic regression model comes out on top with a classification average precision of 62.6%, while the SVM and decision tree models achieve 59.9% and 60.95% average precision, respectively. The logistic regression correctly classifies 235 out of 376 tiles with 62.5% accuracy. Logistic regression analysis of phenotypic attributes can comprehensively predict the risk of BCR in a patient cohort. In cases where recurrence is predicted, the model can also estimate when the BCR is most inclined to occur, facilitating the timely initiation of active surveillance.
These results reaffirm logistic regression as the superior choice for BCR and time relapse prediction when phenotypic features and Gleason scores are utilized as inputs. Therefore, only logistic regression was considered in the following results, as it provided a better classification. The weight assigned to each attribute reflects its significance in predicting recurrence. A higher positive value indicates a major impact on predicting the risk of biochemical recurrence.
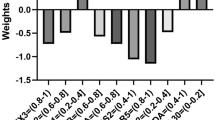
This signature of extracted features and Gleason scores can also be seen in Fig. 10. Notably, the attributes Gleason scores of 8, 9, and 10 are correlated with positive weights, reflecting the correlation between risk of recurrence and cancer aggressiveness. The rest of the attributes with positive weights, such as the value of the maximum ratio contour to whole tiles, number of glands fused, morphology glands means, and the high number of lumina discarded, coincide with juxtaposed, non-circular shaped glands, and clusters of nuclei including former lumina. Negative weighted attributes indicate that tiles presenting BCR enclose a broad number of clusters of nuclei, contours with a high density of nuclei, and glands with small lumina with non-circular morphology.

Phenotypic features and Gleason scores with their respective weights are involved in the logistic regression signature to predict the risk of presenting recurrence
Next, to further enhance BCR prediction, PSA levels were added to the model, already filled with the phenotypic features and Gleason scores. The inclusion of PSA data improved the BCR prediction to a 79.2% accuracy, with a precision of 69.7% for BCR within 24 months and 63.1% for BCR beyond 24 months (Table 4).
4 Discussion
The final logistic model produced an average accuracy of 71% in classifying patients at higher risk of presenting BCR, using available clinical data in cancer diagnosis without additional diagnostic exams or extra computational expenditure. Our findings indicate that adding PSA levels to the extracted phenotypic features and the Gleason score improves the accuracy of BCR prediction. This suggests that when the Gleason score is included as a phenotypic feature, that is not enough to predict patient outcomes.
Prior research [31,32,33], consistently supports the association between more aggressive cancer (higher Gleason scores) and an increased risk of BCR (Fig. 10). Images from patients with a Gleason score of 7 are problematic because it is a transitional category that depends mostly on the predominant pattern [21]. While no explicit distinction was made between Gleason 3 + 4 and Gleason 4 + 3 in this study, this potential oversight was addressed by incorporating morphological attributes independent of the Gleason score, such as glandular structure, lumina organization, and nuclei density. By leveraging these phenotypic features, the model can accurately predict BCR risk for patients with Gleason 3 + 4 or 4 + 3, effectively canceling the need for subgroup differentiation within the Gleason 7 category. This approach allows for a more robust prediction of BCR risk by focusing on histological patterns and tumor morphology, rather than solely relying on Gleason score subcategories.
While Gleason scores have been a predominant focus, our data underscores the value of PSA, especially when aiming for higher accuracy. Although some studies suggest a strong correlation between PSA and tumor aggressiveness (indicated by Gleason scores) [34, 35], contrasting reports suggest that individuals with high Gleason scores but low PSA levels may have pathologic or genetic variants that render them less responsive to current therapies [36]. In this context, our results emphasize PSA as a valuable attribute for high accuracy with high Gleason scores, suggesting that PSA is a dominant predictor. Further tests are required to assess the impact of PSA on the stratification of patients.
Apart from biopsies, PSA tests are conducted when prostate discomfort is expressed. A model integrating phenotypic features, Gleason score, and PSA could enhance accessibility for patients in resource-limited settings, potentially becoming a systematic process in prostate cancer diagnosis, ultimately improving life expectancy. In conjunction with the classifier, we propose a computer-aided detection system for prostate cancer to assist oncologists in their work, optimize the whole H&E slide scanning process, and facilitate the identification of tumorous architecture. Challenges resulting from histological variability and staining differences are addressed through meticulous preprocessing, using a combination of filters to erase artifacts (markers of any color), increase contrast (contrast stretching), and conversion to different color spaces. The mentioned pre-process phase can benefit pathologists by enhancing tissue readability through different organs and improving image quality.
The proposed U-net model provides a significant advantage in the segmentation of prostate adenocarcinoma. It is capable of segmenting glands without visible lumen, which is a feature that has often been overlooked or failed to be segmented by previous models [14, 15, 37], but considering that feature has also been uncommon [13]. Its robustness and ability to be generalized have also been proven by the valid segmentation of elements of different sizes and from multiple sources. The model performance remained high, regardless of the source of the training data.
Implemented DeepLabV3 models showed great results in gastric cancer segmentation [38] but were unfit on invariant stain patterns. Higher segmentation performance can potentially be achieved with Vision Transformers (ViT) algorithms [39], either alone or in combination with a CNN [40]. Nevertheless, current ViT models require large annotated datasets and display insufficient interpretability [41]. The U-net model, with its straightforward implementation and efficient training, demonstrates the substantial potential for transfer learning across various image sources.
Improved U-net models have recently been applied for medical image segmentation [42, 43]. Notably, the U-net method introduced earlier is intelligible for medical professionals [44]. Its manageable and customizable implementation allows for adjusting the kernel size and number of layers, according to the type and source of the sample, reducing the risk of overfitting. This adaptability provides pathology specialists in LA with a wider range of operations. Its highly adaptable architecture can easily be adapted and modified to accommodate tasks and images of different scales while ensuring simple transfer learning integration. Spatial context and subtle details are preserved in highly detailed images, specifically histological images, as a result of the U-net encoder-decoder structure while trainable with limited labeled data. The shallow depth enables its training on machines with restricted computational resources and limited GPU capacity, consequently more feasible, realistic, and manageable for pathologists in LA, compared to more recent architectures.
Increasing the number of tiles of higher Gleason scores and, consequently, the spread of a tumor, may enhance classifier accuracy. This increased predictive power could lead to significant discoveries regarding the origin of recurrence. For instance, elements of the extracellular matrix (ECM) responsible for tissue and architectural structure, along with morphological changes affecting its function, can result in increased proliferation of cancer cells and metastasis development [45]. A strong link between ECM and BCR in prostate cancer had previously been underlined [46], the methodology could therefore benefit from investigating anatomical structure related to ECM and its influence on its structural support to surrounding cells. Although collagen, a predominant ECM component, was not considered in our methodology, it has been emphasized that its features, such as fiber straightness and density, may predict malignant potential in breast cancer [47]. Moreover, there have been advancements in collagen fiber segmentation in WSI [48, 49]. As prostate diagnosis and prognosis mainly rely on gland structure, stroma structures are often overlooked. Considering and collecting collagen changes in the tissue [50] might facilitate the prediction of the time of recurrence. Finally, it may be possible to predict the exact time of recurrence by including more patients with BCR in the investigation, as the TCGA data only included 75 cases of recurrence.
Our study significantly contributes to the existing knowledge in this area. Unlike limited training sets [51], our model was developed using a diverse range of specimens from the cohort, graded from Gleason 5 to 10. We also extracted and examined the percentage of tumor volume as a predictor for early BCR in high-risk patients. Furthermore, our approach goes beyond the Gleason pattern by incorporating information on nuclei density and spatial data as predictors for BCR. Since we used attributes already ubiquitous in clinical practice, we believe this approach holds great promise for improving BCR prediction in clinical settings with limited resources. The proposed solution can significantly shorten the often lengthy waiting times for diagnosis and provide superior histopathological assessments, which are scarce in LA [52]. Computer-aided software (CAD) can alleviate the limited number and unequal distribution of highly-trained healthcare providers [53], thus enhancing BCR detection. Until now, no methodologies have been developed in the literature to provide adequate and evenly distributed access to the technologies required to predict the risk of prostate BCR and its possible treatment in LA [54]. This is consistently restricted by the high cost of cancer treatment and insufficient training [55]. The implementation of the solution can be hindered by the regulation and standard on medical devices set by the country, to ensure patients' data privacy, efficacy, and reliability of the suggested tools. Regular maintenance and updates are required to warrant the accuracy of the models but might be incompatible with current electronic systems used in clinical institutions. Constant training and familiarity of personnel, updates, and integration of the AI-driven solution can strain clinical institutions with limited resources. Priorities may shift away from implementing modern technological solutions and instead favor using established devices.
The limitations of this study are primarily associated with the restricted dataset. Specifically, due to the stringent patient data requirements, including PSA levels, H&E WSI, and Gleason scores, we were compelled to rely solely on the TCGA database for comprehensive information. Unfortunately, this database contained fewer than 80 cases with BCR. Therefore, to strengthen the proposed model, the expansion of the dataset is needed, by incorporating biopsies and clinical information from multiple healthcare centers. Incorporating radical prostatectomy specimens could also improve the predictive power of the model by allowing it to account for spatial variations and tumor distribution across the entire prostate. Furthermore, broadening the spectrum of extracted phenotypic features, particularly those related to histological structures like epithelium or ECM components such as collagen, may bolster our predictive results.
Moreover, the study primarily relies on biopsy characteristics, such as Gleason scores and histological features, without incorporating postoperative pathological characteristics for patients treated with radical prostatectomy. Variables such as surgical margin status, extraprostatic extension, and seminal vesicle invasion are critical for stratifying recurrence risk and predicting overall mortality. Recent evidence has demonstrated that positive surgical margins significantly increase the risk of BCR and prostate cancer-specific mortality, particularly in conjunction with other adverse pathological features such as extraprostatic extension and high Gleason scores [56, 57]. Furthermore, the extent and location of positive surgical margins are also highly prognostic. These findings underscore the importance of integrating pathological data into predictive models, especially for patients treated surgically.
WSI has the potential to stratify patients at risk of BCR during the diagnostic phase or prior to initiating treatment. By identifying patients who may benefit from more aggressive therapies or closer post-treatment monitoring, this approach could significantly optimize treatment strategies. The predictive information provided by the model demonstrates utility both before and after definitive treatment. Pre-treatment, the model’s ability to estimate recurrence risk could inform therapeutic decision-making, particularly in scenarios where treatment choices, such as surgery versus radiotherapy, hinge on recurrence probabilities. Post-treatment, for patients in remission, the model could assist in tailoring follow-up schedules and interventions, potentially facilitating earlier detection of recurrence risks compared to conventional methodologies. Although the development of simplified AI workflows compatible with less expensive hardware is proposed, WSI’s clinical utility remains largely constrained to institutions with the infrastructure necessary to implement this technology.
A method to phenotypically characterize prostate cancer histopathology images assigned Gleason scores 6 up to 10 is presented. We show evidence that specific image features, Gleason scores, and PSA levels, improve the prediction of biochemical recurrence. This method offers a cost-efficient solution for implementation in healthcare centers in rural areas of LA, providing a valuable tool for biochemical recurrence risk prediction.
Data availability
As stated in the Methods Section, the list of the data and images downloaded from public databases are available in Supplementary Materials.
References
Nelson WG, Antonarakis ES, Carter HB, De Marzo AM, DeWeese TL. Prostate cancer. In: Niederhuber JE, Armitage JO, Kastan MB, Doroshow JH, Tepper JE, editors. Abeloff’s clinical oncology. 6th ed. Philadelphia: Elsevier; 2020. p. 1401- 1432.e7. https://doi.org/10.1016/B978-0-323-47674-4.00081-5.
Bae J, Yang SH, Kim A, Kim HG. RNA-based biomarkers for the diagnosis, prognosis, and therapeutic response monitoring of prostate cancer. Urol Oncol. 2022;40(3):105.e1-105.e10. https://doi.org/10.1016/j.urolonc.2021.11.012.
Simon NI, Parker C, Hope TA, Paller CJ. Best approaches and updates for prostate cancer biochemical recurrence. Am Soc Clin Oncol Educ Book Ann Meet. 2022;42:1–8. https://doi.org/10.1200/EDBK_351033.
Babyar J. In search of Pan-American indigenous health and harmony. Global Health. 2019;15:16. https://doi.org/10.1186/s12992-019-0454-1.
Nuche-Berenguer B, Sakellariou D. Socioeconomic determinants of participation in cancer screening in Argentina: a cross-sectional study. Front Pub Health. 2021;9:699108. https://doi.org/10.3389/fpubh.2021.699108.
Chu J, Li N, Gai W. Identification of genes that predict the biochemical recurrence of prostate cancer. Oncol Lett. 2018;16(3):3447–52. https://doi.org/10.3892/ol.2018.9106.
Manceau C, Beauval JB, Lesourd M, Almeras C, Aziza R, Gautier JR, et al. MRI characteristics accurately predict biochemical recurrence after radical prostatectomy. J Clin Med. 2020;9(12):3841. https://doi.org/10.3390/jcm9123841.
Wei C, Zhang Y, Malik H, Zhang X, Alqahtani S, Upreti D, et al. Prediction of postprostatectomy biochemical recurrence using quantitative ultrasound shear wave elastography; imaging. Front Oncol. 2019;9:572. https://doi.org/10.3389/fonc.2019.00572.
Howard FM, Dolezal J, Kochanny S, Khramtsova G, Vickery J, Srisuwananukorn A, et al. Multimodal prediction of breast cancer recurrence assays and risk of recurrence. bioRxiv. 2022. https://doi.org/10.1101/2022.07.07.499039.
Hu W, Li X, Li C, Li R, Jiang T, Sun H, Huang X, Grzegorzek M, Li X. A state-of-the-art survey of artificial neural networks for whole-slide Image analysis: from popular convolutional neural networks to potential visual transformers. Comput Biol Med. 2023;161:107034. https://doi.org/10.1016/j.compbiomed.2023.107034.
Sussman L, Garcia-Robledo JE, Ordóñez-Reyes C, Forero Y, Mosquera AF, Ruíz-Patiño A, et al. Integration of artificial intelligence and precision oncology in Latin America. Front Med Technol. 2022. https://doi.org/10.3389/fmedt.2022.1007822.
Guo J, Li B. The application of medical artificial intelligence technology in rural areas of developing countries. Health Equity. 2018;2(1):174–81. https://doi.org/10.1089/heq.2018.0037.
Noorbakhsh J, Farahmand S, Foroughi Pour A, Namburi S, Caruana D, Rimm D, et al. Deep learning-based cross-classifications reveal conserved spatial behaviors within tumor histological images. Nat Commun. 2020;11(1):6367. https://doi.org/10.1038/s41467-020-20030-5.
Xu Y, Li Y, Wang Y, Liu M, Fan Y, Lai M, Chang EI. Gland instance segmentation using deep multichannel neural networks. IEEE Trans Biomed Eng. 2017;64(12):2901–12. https://doi.org/10.1109/TBME.2017.2686418.
Ren J, Sadimin E, Foran DJ, Qi X. Computer aided analysis of prostate histopathology images to support a refined Gleason grading system. In: Angelini ED, Styner MA, Angelini ED (Eds). Medical Imaging 2017: Image Processing Article 101331V (Progress in Biomedical Optics and Imaging—Proceedings of SPIE; Vol. 10133). SPIE. 2017. https://doi.org/10.1117/12.2253887
Salvi M, Bosco M, Molinaro L, Gambella A, Papotti M, Acharya UR, Molinari F. A hybrid deep learning approach for gland segmentation in prostate histopathological images. Artif Intell Med. 2021;115:102076. https://doi.org/10.1016/j.artmed.2021.102076.
Qiu Y, Hu Y, Kong P, Xie H, Zhang X, Cao J, Wang T, Lei B. Automatic prostate Gleason grading using pyramid semantic parsing network in digital histopathology. Front Oncol. 2022;12:772403. https://doi.org/10.3389/fonc.2022.772403.
Murchan P, Ó’Brien C, O’Connell S, McNevin CS, Baird AM, Sheils O, Ó Broin P, Finn SP. Deep learning of histopathological features for the prediction of tumour molecular genetics. Diagnostics. 2021;11(8):1406. https://doi.org/10.3390/diagnostics11081406.
Saillard C, Schmauch B, Laifa O, Moarii M, Toldo S, Zaslavskiy M, et al. Predicting survival after hepatocellular carcinoma resection using deep learning on histological slides. Hepatology. 2020;72(6):2000–13. https://doi.org/10.1002/hep.31207.
Foroughi Pour A, White BS, Park J, Sheridan TB, Chuang JH. Deep learning features encode interpretable morphologies within histological images. Sci Rep. 2022;12:9428. https://doi.org/10.1038/s41598-022-13541-2.
Sim HG, Telesca D, Culp SH, Ellis WJ, Lange PH, True LD, et al. Tertiary Gleason pattern 5 in Gleason 7 prostate cancer predicts pathological stage and biochemical recurrence. J Urol. 2008;179(5):1775–9. https://doi.org/10.1016/j.juro.2008.01.016.
Deng FM, Donin NM, Pe Benito R, Melamed J, Le Nobin J, Zhou M, et al. Size-adjusted quantitative Gleason score as a predictor of biochemical recurrence after radical prostatectomy. Eur Urol. 2016;70(2):248–53. https://doi.org/10.1016/j.eururo.2015.10.026.
Park SW, Hwang DS, Song WH, Nam JK, Lee HJ, Chung MK. Conditional biochemical recurrence-free survival after radical prostatectomy in patients with high-risk prostate cancer. Prostate Int. 2020;8(4):173–7. https://doi.org/10.1016/j.prnil.2020.07.004.
Pinckaers H, van Ipenburg J, Melamed J, et al. Predicting biochemical recurrence of prostate cancer with artificial intelligence. Commun Med. 2022;2:64. https://doi.org/10.1038/s43856-022-00126-3.
Veta M, Heng YJ, Stathonikos N, Bejnordi BE, Beca F, Wollmann T, et al. Predicting breast tumor proliferation from whole-slide images: The TUPAC16 challenge. Med Image Anal. 2019;54:111–21. https://doi.org/10.1016/j.media.2019.02.012.
Ronneberger O, Fischer P, Brox T. U-Net: convolutional networks for biomedical image segmentation. In: Navab N, Hornegger J, Wells WM, Frangi AF, editors. Medical image computing and computer-assisted intervention—MICCAI 2015. Cham: Springer International Publishing; 2015. p. 234–41.
Gamarra M, Zurek E, Escalante HJ, Hurtado L, San-Juan-Vergara H. Split and Merge Watershed: a two-step method for cell segmentation in fluorescence microscopy images. Biomed Signal Process Control. 2019;53:101575. https://doi.org/10.1016/j.bspc.2019.101575.
Cazals F, Giesen J. Delaunay triangulation based surface reconstruction. In: Boissonnat JD, Teillaud M, editors. Effective computational geometry for curves and surfaces. Berlin, Heidelberg: Springer; 2006. https://doi.org/10.1007/978-3-540-33259-6_6.
Vino G, Sappa AD. Revisiting harris corner detector algorithm: a gradual thresholding approach. In: Kamel M, Campilho A, editors. Image analysis and recognition, vol. 7950. ICIAR 2013. Lecture Notes in Computer Science. Berlin, Heidelberg: Springer; 2013. https://doi.org/10.1007/978-3-642-39094-4_40.
Zhang L, Han C, Cheng Y. 2017 Improved SLIC superpixel generation algorithm and its application in polarimetric SAR images classification. IEEE International geoscience and remote sensing symposium (IGARSS), Fort Worth, TX, USA, 2017, pp. 4578-4581, https://doi.org/10.1109/IGARSS.2017.8128020
Kishan AU, Shaikh T, Wang P-C, Reiter RE, Said J, Raghavan G, et al. Clinical outcomes for patients with Gleason score 9–10 prostate adenocarcinoma treated with radiotherapy or radical prostatectomy: a multi-institutional comparative analysis. Eur Urol. 2017;71(5):766–73. https://doi.org/10.1016/j.eururo.2016.06.046.
Van den Broeck T, van den Bergh RCN, Arfi N, Gross T, Moris L, Briers E, et al. Prognostic value of biochemical recurrence following treatment with curative intent for prostate cancer: a systematic review. Eur Urol. 2019;75(6):967–87. https://doi.org/10.1016/j.eururo.2018.10.011.
Knipper S, Karakiewicz PI, Heinze A, Preisser F, Steuber T, Huland H, Graefen M, Tilki D. Definition of high-risk prostate cancer impacts oncological outcomes after radical prostatectomy. Urol Oncol. 2020;38(4):184–90. https://doi.org/10.1016/j.urolonc.2019.12.014.
Lojanapiwat B, Anutrakulchai W, Chongruksut W, Udomphot C. Correlation and diagnostic performance of the prostate-specific antigen level with the diagnosis, aggressiveness, and bone metastasis of prostate cancer in clinical practice. Prostate Int. 2014;2(3):133–9. https://doi.org/10.12954/PI.14054.
Ngwu PE, Achor GO, Eziefule VU, Orji JI, Alozie FT. Correlation between prostate specific antigen and prostate biopsy Gleason score. Ann Health Res. 2019. https://doi.org/10.30442/ahr.0502-26-56.
Kim DW, Chen MH, Wu J, Huland H, Graefen M, Tilki D, D’Amico AV. Prostate-specific antigen levels of ≤4 and >4 ng/mL and risk of prostate cancer-specific mortality in men with biopsy Gleason score 9 to 10 prostate cancer. Cancer. 2021;127(13):2222–8. https://doi.org/10.1002/cncr.33503.
Singh M, Kalaw EM, Giron DM, Chong KT, Tan CL, Lee HK. Gland segmentation in prostate histopathological images. J Med Imagin. 2017;4(2):027501. https://doi.org/10.1117/1.JMI.4.2.027501.
Sun M, Zhang G, Dang H, Qi X, Zhou X, Chang Q. Accurate gastric cancer segmentation in digital pathology images using deformable convolution and multi-scale embedding networks. IEEE Access. 2019;7:75530–41. https://doi.org/10.1109/ACCESS.2019.2918800.
Ikromjanov K, Bhattacharjee S, Hwang Y-B, Sumon RI, Kim H-C, Choi H-K. Whole slide image analysis and detection of prostate cancer using vision transformers. In 2022 international conference on artificial intelligence in information and communication (ICAIIC). 2022. 399–402. https://doi.org/10.1109/ICAIIC54071.2022.9722635
Zhang Y, Liu H, Hu Q. TransFuse: fusing transformers and CNNs for medical image segmentation. Cham: Springer International Publishing; 2021. https://doi.org/10.48550/arXiv.2102.08005.
Khan RF, Lee BD, Lee MS. Transformers in medical image segmentation: a narrative review. Quant Imagin Med Surg. 2023;13(12):8747–67. https://doi.org/10.21037/qims-23-542.
Li D, Chu X, Cui Y, Zhao J, Zhang K, Yang X. Improved U-Net based on contour prediction for efficient segmentation of rectal cancer. Comput Methods Programs Biomed. 2022;213:106493. https://doi.org/10.1016/j.cmpb.2021.106493.
Sanjar K, Bekhzod O, Kim J, Kim J, Paul A, Kim J. Improved U-Net: fully convolutional network model for skin-lesion segmentation. Appl Sci. 2020. https://doi.org/10.3390/app10103658.
Yin X-X, Sun Le, Fu Y, Lu R, Zhang Y. U-Net-based medical image segmentation. J Healthcare Eng. 2022;2022:1–16. https://doi.org/10.1155/2022/4189781.
Penet M-F, Kakkad S, Pathak AP, Krishnamachary B, Mironchik Y, Raman V, et al. Structure and function of a prostate cancer dissemination-permissive extracellular matrix. Clin Cancer Res. 2017;23:2245–54. https://doi.org/10.1158/1078-0432.CCR-16-15.
Marin L, Casado F. Prediction of prostate cancer biochemical recurrence by using discretization supports the critical contribution of the extracellular matrix genes. Sci Rep. 2023. https://doi.org/10.1038/s41598-023-35821-1.
Conklin MW, Gangnon RE, Sprague BL, Gemert LV, Hampton JM, Eliceiri KW, et al. Collagen alignment as a predictor of recurrence after ductal carcinoma in situ. Cancer Epidemiol, Biomark Prevent. 2018;27(2):138–45. https://doi.org/10.1158/1055-9965.EPI-17-0720.
Jung H, Suloway C, Miao T, Edmondson EF, Morcock DR, Deleage C et al. Integration of deep learning and graph theory for analyzing histopathology whole-slide images. In 2018 IEEE applied imagery pattern recognition workshop (AIPR). 2018. 1–5. https://doi.org/10.1109/AIPR.2018.87074
Kleczek P, Jaworek-Korjakowska J, Gorgon M. A novel method for tissue segmentation in high-resolution H&E-stained histopathological whole-slide images. Comput Med Imagin Gr. 2020;79:101686. https://doi.org/10.1016/j.compmedimag.2019.101686.
Huang J, Zhang L, Wan D, Zhou L, Zheng S, Lin S, Qiao Y. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct Target Ther. 2021;6(1):153. https://doi.org/10.1038/s41392-021-00544-0.
Hansen J, Bianchi M, Sun M, Rink M, Castiglione F, Abdollah F, et al. Percentage of high-grade tumour volume does not meaningfully improve prediction of early biochemical recurrence after radical prostatectomy compared with Gleason score. BJU Int. 2014;113(3):399–407. https://doi.org/10.1111/bju.12424.
Strasser-Weippl K, Chavarri-Guerra Y, Villarreal-Garza C, Bychkovsky BL, Debiasi M, Liedke PE, et al. Progress and remaining challenges for cancer control in Latin America and the Caribbean. Lancet Oncol. 2015;16(14):1405–38. https://doi.org/10.1016/S1470-2045(15)00218-1.
Barrios CH, Werutsky G, Mohar A, Ferrigno AS, Muller BG, Bychkovsky BL, et al. Cancer control in Latin America and the Caribbean: recent advances and opportunities to move forward. Lancet Oncol. 2021;22(11):e474–87. https://doi.org/10.1016/S1470-2045(21)00492-7.
Reis RBD, Alías-Melgar A, Martínez-Cornelio A, Neciosup SP, Sade JP, Santos M, et al. Prostate cancer in Latin America: challenges and recommendations. Cancer Control J Moffitt Cancer Center. 2020;27(1):1073274820915720. https://doi.org/10.1177/1073274820915720.
de Oliveira FHC, de Lorena Sobrinho JE, da Cruz Gouveia Mendes A, Gutman HMS, Filho GJ, Montarroyos UR. Profile of judicialization in access to antineoplastic drugs and their costs: a cross-sectional, descriptive study based on a set of all lawsuits filed between 2016 and 2018 in a state in the Northeast Region of Brazil. BMC Pub Health. 2022;22(1):1824. https://doi.org/10.1186/s12889-022-14199-1.
Pellegrino F, Falagario UG, Knipper S, Martini A, Akre O, Egevad L, et al. ERUS scientific working group on prostate cancer of the European association of urology. assessing the impact of positive surgical margins on mortality in patients who underwent robotic radical prostatectomy: 20 years’ report from the EAU robotic urology section scientific working group. Eur Urol Oncol. 2024;7(4):888–96. https://doi.org/10.1016/j.euo.2023.11.021.
Falagario UG, Knipper S, Pellegrino F, Martini A, Akre O, Egevad L, et al. ERUS scientific working group on prostate cancer of the European association of urology. Prostate cancer-specific and all-cause mortality after robot-assisted radical prostatectomy: 20 years’ report from the European association of urology robotic urology section scientific working group. Eur Urol Oncol. 2024;7(4):705–12. https://doi.org/10.1016/j.euo.2023.08.005.
Author information
Authors and Affiliations
Contributions
L.E.M. and D.I.Z. Wrote the main manuscript. L.E.M and J.I.G. analyzed and interpreted the data. D.R. and F.L.C. designed the work, and critically reviewed the manuscript. All authors approved the manuscript for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Marin, L.E., Zavaleta-Guzman, D.I., Gutierrez-Garcia, J.I. et al. Prediction of biochemical prostate cancer recurrence from any Gleason score using robust tissue structure and clinically available information. Discov Onc 16, 128 (2025). https://doi.org/10.1007/s12672-025-01896-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-025-01896-7